
Schaeffer Center White Paper Series | DOI: 10.25549/q1nw-an61
Policy Context
The purported goal of the Inflation Reduction Act (IRA) of 2022 is to lower healthcare costs for Americans. Whether it will do so remains unclear. Some have argued that “millions of seniors on Medicare will see savings amounting to thousands of dollars every year.”1 Others counter that the IRA will have adverse effects on drug discovery and “will do little or nothing to lower the cost of healthcare.”2 It isn’t clear which view will prevail.
Part of the problem is a lack of detail. In March 2023, the Centers for Medicare and Medicaid Services (CMS) released additional information for the Medicare Drug Price Negotiation program,3 but how CMS will implement a key feature—the “maximum fair price”—remains unclear.
This white paper provides recommendations to mitigate the potential adverse effects of the IRA while preserving incentives for future innovation. We suggest ways to make the price-negotiation process more transparent and tied to drug value and how to address the concern that cost-effectiveness studies discriminate against the disabled or severely ill. Crucial to doing so is to allow drug pricing to change with real-world evidence of value.
Key Takeaways
- The IRA price-negotiation process lacks clarity about how “maximum fair prices” for selected drugs are determined. We recommend price determination be done transparently and linked to value via methods that take into account the preferences of patients—specifically, generalized risk-adjusted cost-effectiveness (GRACE) methodologies—and through collaboration with relevant stakeholders.
- To mitigate the possibility that the IRA price-negotiation provisions will weaken incentives for new indications and post-launch evidence generation, we recommend that CMS implement a process that delays price negotiation when new evidence is established.
- To better allow payers to negotiate prices with drug value, we recommend that manufacturers be exempt from the IRA’s inflation rebate provision for a specified time after launch and follow a three-part pricing schedule that allows prices to increase as new evidence around treatment efficacy, effectiveness and safety is generated; be capped at economic inflation once a value-based price can be determined; and then fall due to either CMS price negotiations or—preferably— significant generic or biosimilar entry into the market.
A press release covering this white paper’s findings is available here.
Abstract
The Inflation Reduction Act of 2022 (IRA) includes several consequential provisions aimed at reducing drug spending and increasing access to pharmaceuticals for millions of Americans. However, the provisions also limit insurers’ ability to implement cost-containment measures and may discourage investments in new drugs and indications. We offer three recommendations to mitigate these potential unintended consequences. First, the calculation of a “maximum fair price” for drugs should be transparent and focus on measured social value rather than price minimization. Second, post-market approval of new indications should be encouraged by delaying the government price-setting process when new indications are approved. Third, the government should exempt manufacturers from inflation rebate penalties if additional information (e.g., real-world evidence, new clinical trial data, or new indication approvals) demonstrates more value in a drug post-approval. Implementing these three strategies would balance the competing goals of incentivizing innovation, increasing patient access and reducing spending.
Introduction
On August 16, 2022, President Joe Biden signed the Inflation Reduction Act (IRA) of 2022 into law. The IRA’s Medicare-related provisions fall into two general categories: (1) reduce prescription drug prices, and (2) reduce beneficiary cost sharing and premiums.4 While these apply to Medicare only, they are likely to ripple throughout the healthcare sector. In this white paper, we suggest policy recommendations to allay some of the unintended consequences of the IRA drug-pricing provisions, which include three key features:
- Requires the government to negotiate prices for certain Medicare-covered, single-source drugs (starting with 10 Part D (i.e., self-administered) drugs in 2026, and expanding to Part B drugs (i.e., administered in a doctor’s office or hospital) by 2028, and accumulating more drugs over time)
- Requires manufacturers to pay rebates to Medicare if prices of single-source drugs in Part B or Part D increase faster than the consumer price index
- Reforms the Medicare Part D benefit, increasing liability among Part D plans and required manufacturer-financed discounts
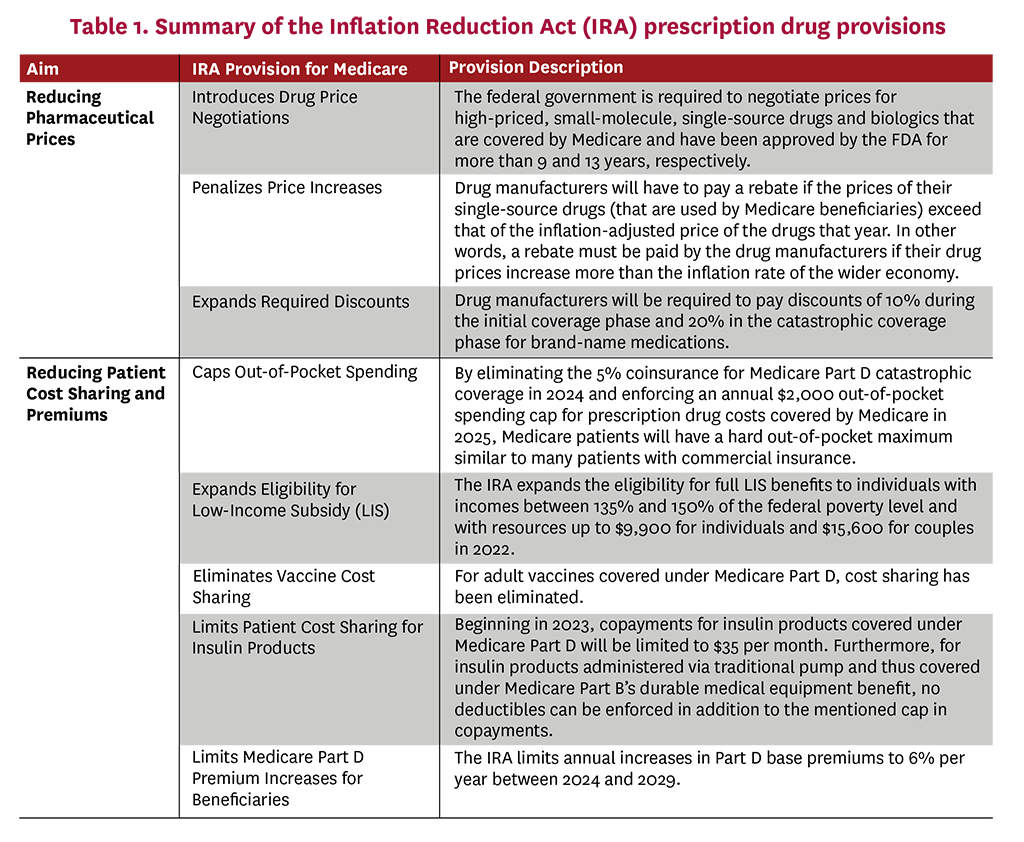
Specifically, the IRA introduces drug-price negotiations by requiring the federal government to negotiate “maximum fair prices” with drug manufacturers for certain brand-name, single-source drugs covered under Medicare Part B and Part D. This provision amends Medicare’s noninterference clause—which prevents the secretary of the U.S. Department of Health & Human Services (HHS) from interfering with negotiations between drug manufacturers, pharmacies and Medicare prescription drug plans—and establishes a new Drug Price Negotiation Program. This program requires HHS to negotiate directly with drug manufacturers on the prices of select pharmaceuticals among the 50 drugs with the highest spending under Medicare Part D and the 50 drugs with the highest spending under Medicare Part B, first effective in 2026 and 2028, respectively.5 The negotiation, however, excludes certain treatments such as those with generic competition, orphan drugs, and small biotech drugs, and only comes into effect nine years (for small-molecule drugs) or 13 years (for biologics) after market approval by the Food and Drug Administration (FDA).6 The penalties for not participating in price negotiation are steep: Manufacturers that do not comply will be subject to excise taxes and civil monetary penalties between 65% and 95% of product sales.
In addition, the IRA also penalizes price increases and expands required discounts on branded, single-source drugs. Specifically, drug manufacturers must pay a rebate to CMS if their prices—the average sales price (ASP) for Part B drugs or the average manufacturer price (AMP) for Part D drugs—increase faster than the general consumer price index for all urban consumers (CPI-U). Notably, AMP does not reflect manufacturer rebates negotiated with pharmacy benefit managers (PBMs) and plans, effectively tying rebate penalties to list rather than net price increases (which are inclusive of the post-sale rebates or discounts manufacturers pay to payers). List prices have already been shown to differ dramatically from and increase faster than net prices—potentially due to recent growth in the size of average discounts/rebates as a share of the list price—and may make it more likely for manufacturers to be subjected to inflation rebate penalties even though their collected net profits will increase slower than the list price increases itself.7-10 Moreover, these inflation rebate penalties are structurally similar to those in effect in the Medicaid program, where they account for the majority of rebates.11
Furthermore, the IRA reforms the structure of the Medicare Part D standard benefit, notably the manufacturer discount program. Once fully phased in, the IRA will require manufacturers to pay 20% discounts on branded drugs in the Part D catastrophic phase and 10% discounts in the initial coverage phase, in addition to expanding these discounts to beneficiaries who receive low-income subsidies (LIS). Compared with the pre-IRA manufacturer coverage gap discount program—whereby manufacturers owed 70% discounts on branded drugs taken by non-LIS beneficiaries in the coverage gap phase—these reforms will increase manufacturer-financed discounts considerably, albeit with significant variation across drug classes.12 The Part D redesign also significantly increases liability for Part D plans in the catastrophic coverage phase by reducing the share of spending paid directly by the federal reinsurance program.
Several IRA provisions will reduce patient cost sharing: instituting an out-of-pocket (OOP) maximum, smoothing patient liability over the year, expanding eligibility for LIS recipients, eliminating vaccine cost sharing, and limiting cost sharing for insulin products. Moreover, the IRA also includes provisions to limit Part D premium increases for beneficiaries. Finally, the IRA further delays the implementation of the Trump Administration’s Rebate Rule to 2032, delaying the effective requirement to share negotiated rebates with patients at the point of sale. In the short run, the IRA’s reduced cost-sharing provisions and price controls should increase access to pharmaceuticals. However, there is compelling evidence that, in the long run, there may be adverse effects for patients due to harmful impacts on life science research and development (R&D) investment decisions.
Adverse Effects of the IRA on Innovation and Plans
Finding 1: The IRA may reduce the discovery of new treatments.
Lowering pharmaceutical revenues leads to less R&D investment and fewer drug discoveries over time.13-15 The IRA is expected to reduce revenue to pharmaceutical manufacturers from the combined effects of drug price negotiation, inflation rebates, and required manufacturer discounts. Taken together, these provisions have been estimated to lead to an approximately 31% decrease in U.S. pharmaceutical revenues through 2039 and result in 135 fewer new drug approvals during the same period.16
Lowered revenues may lead to less research, especially for follow-on drug innovation. One study demonstrated that introduction of the Part D program and thus increased market demand led to an increase in innovation overall, but skewed toward non-breakthrough innovations.17 Thus, the expected reduction in revenues with the IRA is likely to decrease innovation for both novel, groundbreaking drugs as well as those that are less novel but have large consumer markets, such as the elderly population, which accounts for a significant portion of overall healthcare and pharmaceutical drug utilization in this country.17 Consequently, it would not be surprising that potential decreases in Medicare reimbursements due to the IRA’s price control provisions may reduce financial incentives to develop drugs against diseases that disproportionately impact the elderly, such as Alzheimer’s disease, cancer and heart failure.15
According to drug manufacturers, the IRA is already impacting life science companies’ R&D investment decisions. For example, Alnylam mentioned in its October 2022 earnings report that it had suspended development of a treatment for Stargardt disease as a result of needing to “evaluate the impact of the Inflation Reduction Act.”18 In November 2022, Eli Lilly claimed that the IRA was a key reason it ended investments toward developing a drug for certain blood cancers.19 A November–December 2022 survey from the Pharmaceutical Research and Manufacturers of America indicated that 78% of its member companies are expecting to cancel some of their early-state development projects, and 63% are expecting to shift R&D investment focus away from small molecules as a result of the IRA.20
These statements mirror decreased investment in drug innovation in Europe as a result of pharmaceutical price controls. As a result of the UK National Health Service’s drug cost reductions and 24.4% mandatory manufacturer rebate on branded revenues, only 59% of new medications launched between 2012 and 2021 were available in the UK compared to 85% of those medications being available in the U.S.21 For instance, breakthrough therapies for cystic fibrosis were not available for many years in the UK.22 While not all the uncovered drugs in the UK represent significant clinical advances, many of them do. Trends in decreasing investment are seen in other countries as well, including Germany, France, and Italy, and companies such as Bluebird Bio have indicated withdrawals in developing novel gene therapies for rare diseases as a result of these price controls.21
Finding 2: The IRA may reduce discovery of new uses for existing drugs.
New applications of existing drugs often can be efficiently developed since repurposed medications already have built up extensive portfolios of knowledge concerning human pharmacokinetics, bioavailability and toxicology.23 This breadth of information leads to reduced development timelines—typically3-12 years compared to 17 years for new molecules24,25—and development costs that are 85% less than the cost of developing new drugs.26 Governments have also partnered with pharmaceutical firms to explore the potential for repurposing existing drugs to treat new diseases. Examples include the “Discovering New Therapeutic Uses of Existing Molecules” initiative by the National Center for Advancing Translational Sciences in the U.S. and the Medical Research Council partnership with AstraZeneca in the UK.27,28
However, the IRA price negotiation provisions mean that companies may decrease R&D investments in these areas. Global revenue, expected costs and market size influence the amount of money profit-maximizing companies invest in R&D for new drugs. The IRA reduces the net present value of investments in new indications or other Phase IV evidence as it shortens the horizon over which firms can earn returns on those investments.24,25 The result may be less investment in studies that quantify efficacy, safety and value of applications of existing products to new disease areas. If so, price-negotiation policies set by the IRA would result in reduced return on R&D investments since price negotiations would begin nine years (for small molecules drugs) or 13 years (for biologics) after drug approval, even if new indications are identified.
Consider the case of oncology clinical trials. Often, cancer drugs are approved through an expedited approval track using surrogate outcomes. These outcomes—such as progression-free survival or tumor response rate—are correlated with the key long-term outcomes of interest (such as overall survival), but previous research has shown that this correlation is imperfect.29,30 After drug approval, life science firms are often expected to invest additional resources in confirmatory trials to show that their drug also works in the long run. One recent study, however, found that drug manufacturers receive no pricing premium if they conduct a confirmatory trial with a positive result.31 Thus, it should not be surprising that relatively few confirmatory trials using overall survival outcomes are conducted.32 Similarly, a drug price negotiation late in a drug’s life cycle may curtail R&D investments in determining whether a new drug works well in the real world (effectiveness estimates) or for treating other diseases (new indications).33,34
Finding 3: The IRA could reduce generic competition.
Generic manufacturers usually enter pharmaceutical markets after branded counterparts’ patent protections and exclusivity periods have expired. They often pursue the 180-day exclusivity period incentive granted under the Hatch-Waxman Act to the first generic manufacturer to file for FDA approval and demonstrate non-infringement or patent invalidation. Generic entry decreases drug prices between 50% and 90%. For instance, one analysis found that generic drugs that entered the market between 2002 and 2014 reduced drug prices by 51% in the first year, and a 2005 FDA analysis demonstrated that average relative drug price per dose of the branded drug was reduced by nearly 90% with 15 or more generic entrants.35,36
However, the decrease in brand prices due to negotiations could reduce the prices that any generic firm can charge, disincentivizing generics from pursuing the 180-day exclusivity benefit and thus from entering the market. To understand why, consider the following two key facts. First, generic drugs require a sufficiently discounted price relative to the branded drug to attract a large portion of market share away from the branded market, but generic drugs sold at margins that are too low are likely to undermine their profitability. Second, the development of generic drugs involves high upfront costs (albeit much smaller than their branded counterparts) and, accordingly, the level of generic market entry strongly depends on financial incentives such as potential revenue and market size.37,38 To ensure generic manufacturers the latter point, the 180-day exclusivity period was implemented as a strong financial incentive during which no additional generic competition/entry would be allowed and the first generic entrant would be able to capture significant market share to sell its product at relatively high (generic) prices. For instance, in 2001, the Barr Laboratories’ generic version of Prozac had revenues of $366 million during its 180-day exclusivity period—almost 75% of Barr’s revenue for the entire previous year—which subsequently decreased to only $4 million during the next six months, after this “generic exclusivity” was lost and generic competition increased.39 While the IRA’s planned government price negotiation would reduce prices for branded drugs, these reduced branded prices will likely also reduce generics’ pricing advantage relative to Medicare’s negotiated prices. If this results in a scenario where generic manufacturers cannot expect to generate sufficient volume and revenue to justify entering the market,40 the IRA’s price-control provisions could effectively threaten the generic industry’s financial viability. The IRA does, however, attempt to mitigate this generic-entry issue for biosimilars with a provision that provides up to a two-year delay in CMS selection and price negotiations for branded biologics. This delay is granted if there is a “high likelihood” (as determined by the HHS secretary) of a biosimilar being licensed and marketed within two years of the selected drug publication date.
This concern is even more problematic given that the number of generic manufacturers has already contracted in recent years. In fact, 30% to 40% of generic markets are supplied by one manufacturer and current generic market exit rates exceed those of entry.41,42 There is significant uncertainty as to how much the negotiated branded prices will decrease given that the IRA price-control provisions only establish that branded manufacturers must comply with CMS price negotiations with no clear distinction as to how the “maximum fair price” will be established. Accordingly, there are concerns that the IRA price-control provisions could reduce robust generic competition and undermine the drug price reductions produced by other provisions of the legislation.
Finding 4: Inflation rebates may harm plans’ abilities to negotiate prices for drugs with promising but uncertain benefits.
The IRA’s inflation rebate provision, where manufacturers must pay a rebate to CMS if their prices—the ASP for Part B drugs or the AMP for Part D drugs (essentially list prices)—increase faster than inflation, indicated by CPI-U, is designed as if the value of a drug is known with certainty upon its launch and never changes over time; in practice, however, this is rarely the case. More commonly, a drug’s estimated value fluctuates over time as additional information regarding its real-world clinical effectiveness is revealed after approval.29,43 For instance, as a drug enters the market, patients and physicians gain experience using the treatment; effectiveness and safety data are collected.44 Additionally, surrogate endpoints—which are an imperfect measure of efficacy—may be used as clinical trial outcomes.29,30 Further information regarding drug efficacy is often generated after FDA approval through confirmatory trials (e.g., those that assess overall survival) or observatory real-world data studies (i.e., those that estimate treatment effectiveness in the real world). Notably, efforts are already underway from CMS that would reduce Medicare payments for drugs approved under accelerated timelines but before clinical benefit has been confirmed by required confirmatory studies.45
However, the IRA’s inflation rebate limits payers’ ability to negotiate drug prices over time as new evidence accumulates and hamper CMS’s efforts to tie value of a drug to increased evidence of effectiveness. Although manufacturers currently are not able to charge higher prices for completing confirmatory trials or estimating efficacy in the real world,31 drug manufacturers may be willing to accept lower prices for drugs with the understanding that if new evidence shows the drug has higher value, then payers would be willing to pay higher prices. Similarly, if new evidence shows that the drug has lower value, then payers would expect prices to fall. Due to the inflation-rebate provision, however, drug manufacturers are less likely to accept lower launch prices since they know that their ability to increase prices is limited, even if their drug proves highly effective in the real world over time. Moreover, drug manufacturers may have limited incentive to invest in generating new evidence under the IRA pricing framework as they are unlikely to be able to raise their prices even if they show the drug is more effective than that of competitors or has additional indications. These two features mean that payers’ negotiating ability over time is limited due to (1) a reliance on a more limited set of drug evidence, and (2) the evolution of drug prices being based on inflation rather than new information about a drug’s true real-world value. Thus, payers may be faced with increased launch prices since price changes will be based on inflation rates and less on real-world effectiveness and safety information.
Recommendations
Given that implementation of the prescription drug provisions in the IRA is already underway, we recommend three strategies to limit adverse impacts while steering the IRA toward the goals of increased innovation, greater patient access to new medications and lower costs.

Recommendation 1: Bring transparency and a focus on value to the IRA price-determination process.
Any price negotiation for Medicare-covered drugs should be done transparently and linked to assessments on how much value a drug provides relative to its costs. To assess value, economists commonly use cost-effectiveness analysis (CEA), in which health gains are valued equally regardless of patient disease severity. However, the Affordable Care Act (ACA) and the IRA have prohibited the use of traditional, quality-adjusted life-year-based CEA, as it assigns less value to life extensions of disabled patients as compared to non-disabled or healthier patients. Recent advances in value assessment, however, provide the federal government with a better path forward. Generalized risk-adjusted cost-effectiveness (GRACE) can account for the fact that people value health gains most when facing very poor quality of life (e.g., individuals who are severely ill or disabled), and value health gains less when quality of life is higher.46,47 GRACE responds to prior calls by health economists for broader notions of societal value in assessing medicines.48 GRACE could also be combined with other approaches, such as equal value of life years gained.49
Appropriately linking prices to value incentivizes innovation. The goal of value-based pricing is not to minimize government spending or slash drug prices indiscriminately, but rather to reduce prices for drugs that fail to improve patient well-being while rewarding development of treatments that provide the highest health benefits. For instance, Sovaldi (sofosbuvir)—a drug that cures the hepatitis C virus (HCV)—was priced at $84,000 for one treatment course upon approval in 2013 and was determined by a number of studies to be highly cost effective.50,51 If cost minimization was the only goal, millions of patients with HCV would not have received treatment.
Failing to link drug prices to value could have long-term complications on beneficiary access. Given that the IRA’s price negotiation and inflation restrictions are imposed on drug treatments that target diseases disproportionately impacting the elderly (such as macular degeneration and heart disease), an absence of value-based pricing may lead pharmaceutical companies to reduce R&D for these treatments as they become less profitable. But by linking prices to value, it is possible that innovation could shift more toward interventions that would bring the most value to patients.
The government can take several steps to link prices to value while ensuring patient affordability and innovation.
First, in addition to supporting the existing health technology assessments (HTAs) by private entities, the U.S. should implement a publicly funded HTA-coordinating entity—coined as the Institute for Health Technology Assessment (IHTA)—that would conduct its own HTAs while coordinating and evaluating the quality of privately conducted HTAs.52 In terms of drug pricing, this coordinating entity could partner with established organizations, such as the Institute for Clinical and Economic Review (ICER) and the Agency for Healthcare Research and Quality’s Evidence-based Practice Center, that allow for effective evaluations of newly approved drugs (and poorly studied health interventions) that CMS could utilize to better advise and bring transparency to the price-negotiation process with manufacturers.
Second, it would be irresponsible for the IRA’s price-control provisions—which are currently structured more as a price-setting process than a true negotiation—to set prices without knowing the real value of its covered drugs. Following insights from Lakdawalla and colleagues,52 we suggest that multiple stakeholders—including patient and healthcare consumer organizations, healthcare providers, public/private payers, employers and the drug industry—be brought together to promote unbiased, representative value measurements and shift the determination of the “maximum fair price” to more of a negotiation process. For example, private Part D plans and PBMs already utilize proprietary HTA processes to negotiate lower net drug prices with pharmaceutical companies. Accordingly, in addition to the other mentioned stakeholders, these private payers can have a role in the IHTA’s value assessment process, which CMS can use to better inform its value-based price-negotiation process with manufacturers.
While these recommendations will better link prices to value, there is a possibility that using GRACE to assess treatment value may in certain cases suggest “maximum fair prices” above the upper limits outlined in the IRA, especially for drugs that typically benefit patients suffering from severe diseases. In this event, CMS would not be able to negotiate prices to value based on GRACE per the “maximum fair price” regulations set forth by the IRA. However, utilization of such frameworks would highlight to CMS the value that certain drugs can bring to beneficiaries and, accordingly, additional measures—such as raising the thresholds for highly valuable drugs, as was done by the National Institute for Health and Care Excellence (NICE) and the Cancer Drugs Fund for end-of-life drugs—could be taken to provide patient accessibility while incentivizing innovation for these therapeutically beneficial drugs.53
Recommendation 2: Incentivize the production of new information about effectiveness.
To incentivize investment in understanding whether pharmaceuticals can benefit new patient populations, the price-negotiation period should be delayed beyond the nine-year (for small-molecule drugs) or 13-year (for biologics) time frame when new indications are approved. Many drugs have accrued new indications over a period of years. For example, Humira (adalimumab) was approved by the FDA for sale in 2002 for the treatment of rheumatoid arthritis. By 2021, however, the drug had 11 more indications for diseases ranging from Crohn’s disease to ankylosing spondylitis, with four indications approved 13 years after first approval and the latest indication approved in 2021.54,55 Keytruda (pembrolizumab) was approved in 2014 to treat melanoma, but now is approved to treat 20 different types of cancer.56 These are just two of many examples of drugs that have helped numerous patients beyond their original indication. While IRA price negotiation will not completely eliminate the pursuit of new indications, pending price negotiation will decrease the quantity of R&D funds invested in research to find new indications for existing drugs relative to the pre-IRA status quo.
If IRA negotiations drop drug prices to near generic levels at year nine or 13, patient health could worsen due to lack of incentive to pursue new indications, or patient safety may be harmed due to inappropriate off-label use, compared to the counterfactual where there was no drug price negotiation and thus incentives to conduct additional research. To repurpose already-approved drugs and gain FDA approval for new indications, pharmaceutical firms must invest in clinical trials to show effectiveness and safety of the indication. Firms could have limited ability to recoup their investments in clinical trials for new indications if the IRA’s price negotiation does not permit an extension of market exclusivity. Although physicians could still prescribe these drugs off label, studies have found that off-label use of drugs lacking strong scientific evidence has adverse events rates more than 50% higher than drugs used on label.57
To properly incentivize life science firms to invest in clinical trials for new indications, CMS should implement a process that delays price negotiation when valuable new evidence—particularly evidence of new indication approvals—is created. The duration over which price negotiation is delayed for new evidence and new indications should be implemented by CMS through a transparent process that could be linked to the value of any new indication.
Recommendation 3: Allow exceptions to IRA inflation rebates when new evidence is acquired.
The government should provide exemptions to the inflation-rebate provision during the period immediately after a drug’s launch—during which real-world evidence will accumulate and provide additional information on the drug’s effectiveness—and when new evidence is made available after this initial period. This approach would incentivize drug manufacturers to lower launch prices with the understanding that prices could rise above inflation if new evidence with respect to treatment value became available.
Rather than using top-down government price controls, we recommend that the IRA implement a more flexible approach along the lines of a three-part pricing framework.58 Under this framework, drugs first undergo an initial “evaluation phase” in which manufacturers launch their drug with a low price with the incentive to generate new evidence around treatment efficacy, effectiveness and safety over a period of time. In the UK, for instance, NICE may approve a treatment for a more restricted set of conditions until additional, more robust evidence is generated.59 However, using a low launch price would improve uptake and access to the drug by patients in the short term, and would also accelerate the rate of real-world evidence regarding the drug’s effectiveness. During a subsequent “reward phase,” the drug’s price would reflect the degree to which new evidence has or has not demonstrated changes to the initial estimates of treatment safety and effectiveness. Finally, the “access phase” would utilize robust generic competition to discount branded prices upon its loss of exclusivity, accomplishing the IRA’s intended goal for lower drug prices and improved patient access in the long term.
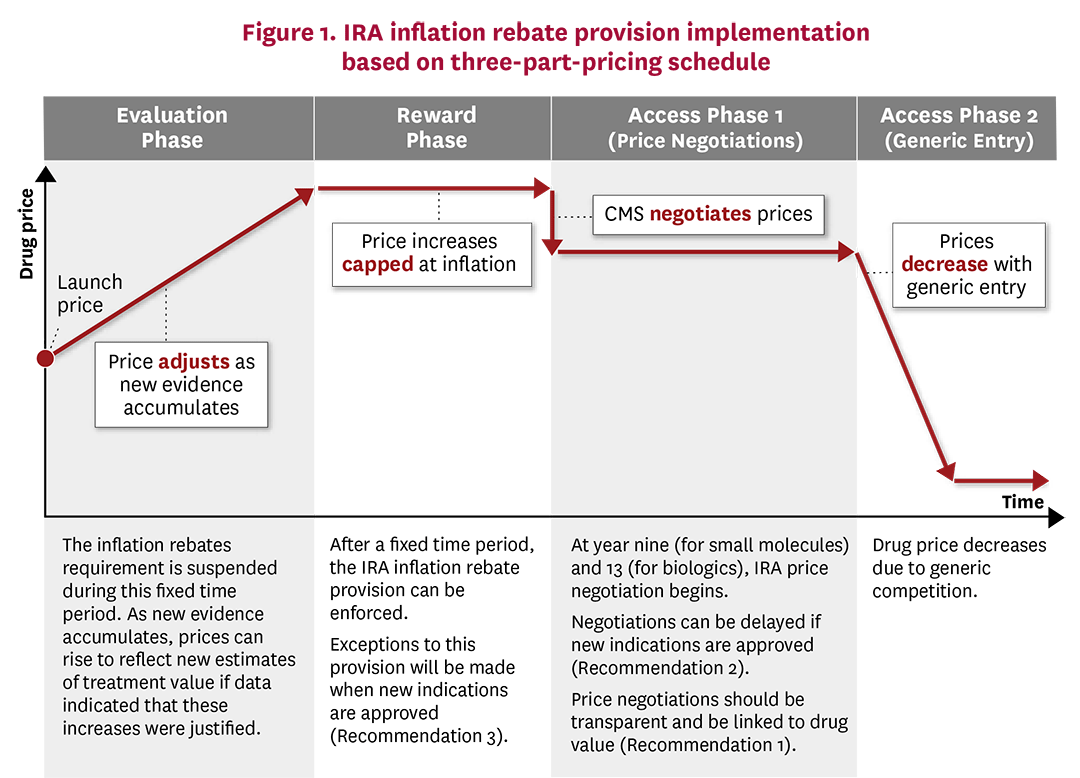
Modifications to the IRA inflation rebate could be readily made so that CMS drug pricing more closely follows the three-part pricing framework (Figure 1). During the initial “evaluation phase,” drug manufacturers would be exempt from the inflation rebate and could increase prices if new clinical trial and real-world data indicated that these price increases were justified. The exact length of the “evaluation phase” will depend on the degree of uncertainty new drugs have at launch, where drugs with more certain benefits would be in this phase for a shorter time frame than more uncertain drugs, as the former would be able to increase prices faster with justified evidence. Once sufficient evidence was accumulated, CMS, Part D Plans and the drug manufacturers would have a stronger idea of the drug’s value and its corresponding price. During the “reward phase,” the IRA inflation rebate could come into effect whereby price increases would be capped at overall economic inflation levels. Finally, during the “access phase,” prices would fall due to either CMS price negotiations or, preferably, significant generic entry into the market.
Other institutions already recognize that treatment value and prices should evolve over time. For instance, ICER exempts treatments from its “unfair price increase” label when new clinical evidence is produced. Similarly, during the “evaluation phase,” prices could adjust in a more market-oriented manner based on the evidence that accumulates. IRA provisions should be made more flexible to better reflect that estimated treatment value evolves over time as evidence accumulates.
Conclusion
Absent reform, the IRA may result in a decline in new drug innovation as well as a decline in research on new indications and evidence generation for long-term effectiveness and safety outcomes. Given that the IRA is the law of the land and its implementation has already begun, our three recommendations steer the potential effects of the IRA toward its goal of improving patient access while encouraging innovation. First, we recommend that any government price-determination process be transparent and focus on value rather than cost minimization. CMS should fund generation of evidence on treatment cost-effectiveness and collaborate with stakeholders to help determine how best to measure treatment value and set a “fair” price. Second, we recommend that innovators be granted delays in the start of the price-setting period when new indications are approved to incentivize research on new indications. Third, we recommend that CMS follows the principles of the three-part pricing schedule and provide exemptions in the inflation-rebate provision for a set period after the initial drug launch. Allowing more flexible pricing during this “evaluation period” could lower drug launch prices while providing appropriate incentives for drug manufacturers to conduct confirmatory clinical trials and collect real-world evidence to demonstrate a drug’s value. With these three steps, we aim to balance the IRA’s goals of incentivizing innovation, increasing access and reducing cost.
The Schaeffer Center White Paper Series is published by the Leonard D. Schaeffer Center for Health Policy & Economics at the University of Southern California. Papers published in this series undergo a rigorous peer review process, led by the Director of Quality Assurance at the USC Schaeffer Center. This process includes external review by at least two scholars not affiliated with the Center. Support for this white paper was provided by the USC Leonard D. Schaeffer Center for Health Policy & Economics. The Schaeffer Center is funded by foundations, corporations, government agencies, individuals and an endowment; a complete list of sponsors can be found in our annual reports (available here). The views expressed herein are those of the authors and do not represent the views of the Schaeffer Center, its sponsors, or FTI Consulting. Disclosures reported by authors are available here.
References
- Conti, R. M., L. Dach and R. Frank. (2022). People Are Underestimating the Significance of Drug Pricing Provisions in Inflation Reduction Act. The Hill, October 6. https://thehill.com/opinion/congress-blog/3676791-people-are-underestimating-the-significance-of-drug-pricing-provisions-in-inflation-reduction-act.
- Hooper, C. L., and D. R. Henderson. (2022). Expensive Prescription Drugs Are a Bargain: The Inflation Reduction Act Gives the Government the Ability to ‘Negotiate’ Prices. People Will Die. Wall Street Journal, September 13. https://www.wsj.com/articles/expensive-medications-are-a-bargain-drug-cost-expense-inflation-reduction-act-brand-name-pharmaceutical-treatments-11663080540.
- Seshamani, M. (2023). Medicare Drug Price Negotiation Program: Initial Memorandum, Implementation of Sections 1191–1198 of the Social Security Act for Initial Price Applicability Year 2026, and Solicitation of Comments. CMS, March 15. https://www.cms.gov/files/document/medicare-drug-price-negotiation-program-initial-guidance.pdf.
- Centers for Medicare and Medicaid Services. (2022) The Inflation Reduction Act Lowers Health Care Costs for Millions of Americans. October 5. https://www.cms.gov/newsroom/fact-sheets/inflation-reduction-act-lowers-health-care-costs-millions-americans.
- Social Security Administration. (n.d.). Subpart 2—Prescription Drug Plans; PDP Sponsors; Financing, Sec. 1860D-11. [42 U.S.C. 1395w-111]. https://www.ssa.gov/OP_Home/ssact/title18/1860D-11.htm.
- Kaiser Family Foundation. (2022). What Are the Prescription Drug Provisions in the Inflation Reduction Act? Accessed October 6, 2022. https://www.kff.org/slideshow/what-are-the-prescription-drug-provisions-in-the-inflation-reduction-act.
- Scott, C., E. Sabot and K. Kirk. (2020). The Myth of Skyrocketing Drug Prices: A Closer Look at the US Gross to Net Problem. PharmaExec.com, May 31. https://www.pharmexec.com/view/myth-skyrocketing-drug-prices-closer-look-us-gross-net-problem.
- Sood, N., T. Shih, K. Van Nuys and D. Goldman. (2017). The Flow of Money Through the Pharmaceutical Distribution System. USC Schaeffer Center White Paper. https://healthpolicy.usc.edu/wp-content/uploads/2017/06/The-Flow-of-Money-Through-the-Pharmaceutical-Distribution-System_Final_Spreadsheet.pdf.
- Sarpatwari, A., F. A. Tessema, M. Zakarian, M. N. Najafzadeh and A. S. Kesselheim. (2021). Diabetes Drugs: List Price Increases Were Not Always Reflected in Net Price; Impact of Brand Competition Unclear. Health Affairs, 40 (5): 772-78. https://www.healthaffairs.org/doi/10.1377/hlthaff.2020.01436.
- Dubois, R. W. (2019). Rx Drug Costs: List Prices Versus Net Prices and the Importance of Staying Within the Data. Health Affairs, March 13. https://www.healthaffairs.org/do/10.1377/forefront.20190312.446522/full/.
- Levinson, D. R. (2015). Medicaid Rebates for Brand-Name Drugs Exceeded Part D Rebates by a Substantial Margin. Department of Health and Human Services, Office of Inspector General, April 21. https://oig.hhs.gov/oei/reports/oei-03-13-00650.pdf.
- Trish, E., K. M. Kaiser, J. Celestin and G. Joyce. (2022). Reforming the Medicare Part D Benefit Design: Financial Implications for Beneficiaries, Private Plans, Drug Manufacturers and the Federal Government. Journal of Health Politics, Policy and Law, 47 (6): 853-77.
- Acemoglu, D., and J. Linn. (2004). Market Size in Innovation: Theory and Evidence From the Pharmaceutical Industry. Quarterly Journal of Economics, 119 (3): 1049-90.
- Dubois, P., O. De Mouzon, F. Scott-Morton and P. Seabright. (2015). Market Size and Pharmaceutical Innovation. RAND Journal of Economics, 46 (4): 844-71.
- Blume-Kohout, M. E., and N. Sood. (2013). Market Size and Innovation: Effects of Medicare Part D on Pharmaceutical Research and Development. Journal of Public Economics, 97: 327-36.
- Philipson, T. J., and T. Durie. (2021). Issue Brief: The Impact of HR 5376 on Biopharmaceutical Innovation and Patient Health. University of Chicago, November 29. https://cpb-us-w2.wpmucdn.com/voices.uchicago.edu/dist/d/3128/files/2021/08/Issue-Brief-Drug-Pricing-in-HR-5376-11.30.pdf.
- Dranove, D., C. Garthwaite and M. Hermosilla. (2022). Does Consumer Demand Pull Scientifically Novel Drug Innovation? RAND Journal of Economics, 53 (3): 590-638.
- Grogan, J. (2022). The Inflation Reduction Act Is Already Killing Potential Cures. Wall Street Journal, November 3. https://www.wsj.com/articles/the-inflation-reduction-act-killing-potential-cures-pharmaceutical-companies-treatment-patients-drugs-prescriptions-ira-manufacturers-11667508291.
- Gelman, M. (2022). Updated: Eli Lilly blames Biden’s IRA for Cancer Drug Discontinuation as the New Pharma Playbook Takes Shape. EndPoints News, November 1.
- Longo, N. (2023). WTAS: Inflation Reduction Act Already Impacting R&D Decisions. PhRMA Foundation, January 17. https://catalyst.phrma.org/wtas-inflation-reduction-act-already-impacting-rd-decisions.
- Board, T. E. (2023). The West’s Drug-Price Self-Sabotage. Wall Street Journal, January 23. https://www.wsj.com/articles/the-wests-drug-self-sabotage-europe-pharmaceutical-investment-price-controls-treatments-covid-cancer-pfizer-11674409032?mod=hp_opin_pos_1.
- Gulland, A. (2016). Cystic Fibrosis Drug Is Not Cost Effective, Says NICE. BMJ, 353: i3409.
- Islam, S., S. Wang, N. Bowden, J. Martin and R. Head. (2022). Repurposing Existing Therapeutics, Its Importance in Oncology Drug Development: Kinases as a Potential Target. British Journal of Clinical Pharmacology, 88 (1): 64-74.
- Pantziarka, P., G. Bouche, L. Meheus, V. Sukhatme, V. P. Sukhatme and P. Vikas. (2014). The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience, 8: 442.
- McCabe, B., F. Liberante and K. I. Mills. (2015). Repurposing Medicinal Compounds for Blood Cancer Treatment. Annals of Hematology, 94 (8): 1267-76.
- Kauppi, D. M. (2015). Therapeutic Drug Repurposing, Repositioning and Rescue. Drug Discovery, 16: 16.
- Pushpakom, S., F. Iorio, P. A. Eyers et al. (2019). Drug Repurposing: Progress, Challenges and Recommendations. Nature Reviews Drug Discovery, 18 (1): 41-58.
- Bertolini, F., V. P. Sukhatme and G. Bouche. (2015). Drug Repurposing in Oncology—Patient and Health Systems Opportunities. Nature Reviews Clinical Oncology, 12 (12): 732-42.
- Shafrin, J., R. Brookmeyer, D. Peneva et al. (2016). The Value of Surrogate Endpoints for Predicting Real-World Survival Across Five Cancer Types. Current Medical Research and Opinion, 32 (4): 731-39.
- Lakdawalla, D. N., J. Shafrin, N. Hou et al. (2017). Predicting Real-World Effectiveness of Cancer Therapies Using Overall Survival and Progression-Free Survival from Clinical Trials: Empirical Evidence for the ASCO Value Framework. Value in Health, 20 (7): 866-75.
- Frank, R. G., M. Shahzad and E. J. Emanuel. (2022). Accelerated Approval of Cancer Drugs: No Economic Reward for Drug Makers That Conduct Confirmatory Trials. Health Affairs, 41 (9): 1273-80.
- Gyawali, B., S. P. Hey and A. S. Kesselheim. (2019). Assessment of the Clinical Benefit of Cancer Drugs Receiving Accelerated Approval. JAMA Internal Medicine, 179 (7): 906-13.
- Huang Bartlett, C., J. Mardekian, M. J. Cotte et al. (2020). Concordance of Real-World Versus Conventional Progression-Free Survival From a Phase 3 Trial of Endocrine Therapy as First-Line Treatment for Metastatic Breast Cancer. (2020). PLoS One, 15 (4): e0227256.
- Fralick, M., A. S. Kesselheim, J. Avorn and S. Schneeweiss. (2018). Use of Health Care Databases to Support Supplemental Indications of Approved Medications. JAMA Internal Medicine, 178 (1): 55-63.
- Gupta, R., N. D. Shah and J. S. Ross. (2019). Generic Drugs in the United States: Policies to Address Pricing and Competition. Clinical Pharmacology & Therapeutics, 105 (2): 329-37.
- Conrad, R., and R. Lutter. (2019). Generic Competition and Drug Prices: New Evidence Linking Greater Generic Competition and Lower Generic Drug Prices. US Food and Drug Administration. https://www.fda.gov/media/133509/download.
- Reiffen, D., and M. R. Ward. (2005). Generic Drug Industry Dynamics. Review of Economics and Statistics, 87 (1): 37-49.
- Scott Morton, F. M. (1997). Entry Decisions in the Generic Pharmaceutical Industry. National Bureau of Economic Research. https://www.nber.org/papers/w6190.
- Harris, G., and J. Slater. (2003). ‘Bitter Pill’: Branded Generics Eat Into Drug Makers’ Profits. Wall Street Journal, April 17. https://www.wsj.com/articles/SB10505264915494100.
- Kfoury, J., A. Guth, J. Mackey and H. Tessler. (2022). How the Inflation Reduction Act Will Impact the Biopharmaceutical Industry. Life Sciences & Pharma: Executive Insights, LEK Insights, XXIV (39). https://www.lek.com/insights/ei/how-inflation-reduction-act-will-impact-biopharmaceutical-industry.
- Berndt, E. R., R. M. Conti and S. J. Murphy. (2017). The Landscape of US Generic Prescription Drug Markets, 2004-2016. National Bureau of Economic Research. https://www.nber.org/papers/w23640.
- Socal, M. P., K. Ahn, J. A. Greene and G. F. Anderson. (2023). Competition and Vulnerabilities in the Global Supply Chain for US Generic Active Pharmaceutical Ingredients: Study Examines the Competition and Vulnerabilities in the global supply chain for US generic active pharmaceutical ingredients. Health Affairs, 42 (3).
- Johannes, C. B., D. C. Beachler, J. B. Layton et al. (2022). Post-Authorization Safety Study of Hospitalization for Acute Kidney Injury in Patients with Type 2 Diabetes Exposed to Dapagliflozin in a Real-World Setting. Drug Safety, 46 (2): 157-74.
- Dang, A. (2023). Real-World Evidence: A Primer. Pharmaceutical Medicine, 37 (1): 25-36.
- Centers for Medicare and Medicaid Services. (2023). HHS Secretary Responds to the President’s Executive Order on Drug Prices. February 14. https://www.cms.gov/newsroom/press-releases/hhs-secretary-responds-presidents-executive-order-drug-prices.
- Lakdawalla, D. N., and C. E. Phelps. (2020). Health Technology Assessment With Risk Aversion in Health. Journal of Health Economics, 72: 102346.
- Lakdawalla, D. N., and C. E. Phelps. (2022). A Guide to Extending and Implementing Generalized Risk-Adjusted Cost-Effectiveness (GRACE). European Journal of Health Economics, 23 (3): 433-51.
- Lakdawalla, D. N., J. A. Doshi, L. P. Garrison Jr., C. E. Phelps, A. Basu and P. M. Danzon. (2018). Defining Elements of Value in Health Care—A Health Economics Approach: An ISPOR Special Task Force Report [3]. Value in Health., 21 (2): 131-39.
- O’Day, K., and D. J. Mezzio. (2021). Demystifying ICER’s Equal Value of Life Years Gained Metric. Value & Outcomes Spotlight, 7 (1): 26-28.
- Linas, B. P., D. M. Barter, J. R. Morgan et al. (2015). The Cost-Effectiveness of Sofosbuvir-Based Regimens for Treatment of Hepatitis C Virus Genotype 2 or 3 Infection. Annals of Internal Medicine, 162 (9): 619-29.
- Chhatwal, J., F. Kanwal, M. S. Roberts and M. A. Dunn. (2015). Cost-Effectiveness and Budget Impact of Hepatitis C Virus Treatment With Sofosbuvir and Ledipasvir in the United States. Annals of Internal Medicine, 162 (6): 397-406.
- Lakdawalla, D., P. Neumann and G. Wilensky. (2021). Health Technology Assessment in the US—A Vision for the Future. USC Schaeffer Center White Paper. https://healthpolicy.usc.edu/research/health-technology-assessment-in-the-u-s-a-vision-for-the-future.
- Baker, R., H. Mason and N. McHugh. (2018). UK Spends Generously to Extend Lives of People With Terminal Illnesses—Against the Public’s Wishes. The Coversation, May 31. https://theconversation.com/uk-spends-generously-to-extend-lives-of-people-with-terminal-illnesses-against-the-publics-wishes-96562.
- Abbvie. (2023). HUMIRA® (Adalimumab) Injection: Prescribing Information. Accessed January 17, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125057s410lbl.pdf.
- Stewart, J. (2022). Humira FDA Approval History. Drugs.com, August 25. https://www.drugs.com/history/humira.html.
- Merck & Co. (2014). KEYTRUDA® (Pembrolizumab) Injection: Prescribing Information. Accessed March 21, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125514s110lbl.pdf.
- Eguale, T., D. L. Buckeridge, A. Verma et al. (2016). Association of Off-Label Drug Use and Adverse Drug Events in an Adult Population. JAMA Internal Medicine, 176 (1): 55-63.
- Goldman, D. P., K. Van Nuys, W-H Cheng et al. (2018). A New Model for Pricing Drugs of Uncertain Efficacy. NEJM Catalyst, 4 (6).
- The Kings Fund. (2023). Access to New Medicines in the English NHS. Accessed March 19, 2023. https://www.kingsfund.org.uk/publications/access-new-medicines-english-nhs.



You must be logged in to post a comment.